
ジェネリック医薬品Q&A
厚生労働省作成「ジェネリック医薬品Q&A」もご参考にしてください。
ジェネリック医薬品への疑問に答えます ~ジェネリック医薬品Q&A~(厚生労働省のサイト)
質問タイトルをクリックいただくと回答が表示されます。
-
ジェネリック医薬品はなぜ安価なのか?
ジェネリック医薬品は先発医薬品と比べて、研究開発に要する期間や費用が少なくてすむため安価になっていますが、品質は先発医薬品と比較して劣ることはありません。また同等の有効性・安全性を有します。
解説通常新薬は9~17年の開発期間と数百億円以上の開発費が必要といわれています。ジェネリック医薬品は先発医薬品と同じ有効成分を使用し、先発医薬品の長年にわたる臨床使用経験等を踏まえて開発、製造されるため、先発医薬品で行われた毒性試験や薬理試験及び臨床試験等の実施の必要はありません。このため3~4年の開発期間と数億円の開発費(但し、最近ではもっと費用が掛かるものが増えています)ですむため、薬価が低く設定されて発売されます。
薬価が低いからといって、品質、有効性、安全性が劣ることはありません。 -
ジェネリック医薬品の原薬は海外の粗悪なものを使っているのではないか?
ジェネリック医薬品に用いる原薬の品質は、国の規制により先発医薬品と同じ品質基準が求められています。例え原薬が海外で製造されたものであっても、品質が劣る粗悪な原薬を用いた製剤が、ジェネリック医薬品として承認・販売されることはありません。
解説ジェネリック医薬品は、原薬及び製剤の品質が先発医薬品の品質と同等あるいはそれ以上でないと承認されません。
例えば原薬の純度に関する品質基準は、日米EU医薬品規制調和国際会議(ICH)の合意に基づく「新有効成分含有医薬品のうち原薬の不純物に関するガイドライン」をジェネリック医薬品についてもそのまま準用しており、製造元が国内、海外とは関係なく、先発医薬品と同じ基準が求められています。
また原薬の海外製造元に対しても、企業及びPMDA(医薬品医療機器総合機構)による定期的な調査を行い、問題の無いことを確認しています。
なお海外からの輸入原薬は、ジェネリック医薬品だけではなく、先発医薬品でも使用されています。 -
ジェネリック医薬品と先発医薬品は添加剤が違う場合があるので、有効性・安全性が同じと言えないのではないか?
医薬品に使用される添加剤は、薬理作用を発揮したり、有効成分の効力に影響を与えたりするようなものは使用できません。ジェネリック医薬品は先発医薬品と添加剤が異なっても有効性・安全性に差が無いことを確認しています。
解説ジェネリック医薬品に使用される添加剤については、先発医薬品と同じ添加剤を使用することが要求されているわけではありません。したがって、添加剤については異なっている場合があります。これはアメリカ等の諸外国においても同様です。
添加剤は、その投与量において薬理作用を発揮したり、有効成分の治療効果を妨げたりするものは使用できません(「日本薬局方製剤総則」)。したがって、医薬品として使用前例のある、安全性が確認されている添加剤が使用されています。使用前例の無い添加剤を使用する場合は、毒性試験等によりその添加剤の安全性を確認したうえでなければ使用できません。
添加剤の成分や配合量が先発医薬品と異なっていても、生物学的同等性試験の実施によって先発医薬品と有効性や安全性に差がないことを確認しています。
このことは、先発医薬品が既承認製剤の添加剤を変更する場合(普通錠に口腔内崩壊錠を追加する場合等)も同様であり、ジェネリック医薬品と同様の試験方法で既承認製剤との生物学的同等性を確認し承認されています。
なお、患者さんの体質によっては、添加剤が原因でアレルギー反応などの副作用等を引き起こすことがまれにありますが、これは、先発医薬品であってもジェネリック医薬品であっても、同様に起こりうることです。そのためその医薬品に含有される添加剤は原則全て、添付文書に記載することとされています。 -
ジェネリック医薬品の製造・品質管理は十分におこなわれているの?
全ての医薬品(先発医薬品、ジェネリック医薬品を問わず)は、共通の省令である、GMP(「医薬品及び医薬部外品の製造管理及び品質管理の基準」)に適合した工場でのみ製造が許可されており、厳格な製造及び品質管理のもと製造されています。
解説医薬品が製造販売承認を得るためには、その製造所での製造がGMP(医薬品及び医薬部外品の製造管理及び品質管理に関する基準)に適合していなければなりません。先発医薬品メーカー、ジェネリック医薬品メーカーを問わず、全ての医薬品は、共通のGMP等の基準を満たした製造所でのみ製造が許されていることになります。
製造された医薬品は、医薬品そのものの品質確認(検査)及びその医薬品の製造記録の照査を行い、その医薬品が適切に製造されたか、品質に問題はないかを確認した後に出荷されます。また、製造された医薬品の一部は保管され、使用期限内に品質が担保されていることを定期的に確認しています。
なお、GMP等の基準の遵守状況についても、医薬品医療機器総合機構(PMDA)及び各都道府県による定期及び不定期の査察により、チェックがなされています。
また、「ジェネリック医薬品の安心使用促進アクションプログラム」の一環として、厚生労働省は都道府県等と協力し、医薬品等一斉監視指導を行っています。この一斉監視指導では、市場に流通しているジェネリック医薬品の品質確認を目的に、行政が実際にジェネリック医薬品を入手し、溶出試験などの品質確認(検査)を行っています。この一斉監視指導の結果は年度ごとに取りまとめられ、厚生労働省のサイトで公表されています。
平成28年度「後発医薬品品質確保対策事業」検査結果報告書について(厚生労働省のサイト)
また文献等において品質に問題があると指摘されたジェネリック医薬品については国立医薬品食品衛生研究所に設けられた「ジェネリック医薬品品質情報検討会」において検討され、適宜品質に関する試験を実施してその結果を公表しています。
ジェネリック医薬品品質情報検討会(国立医薬品食品衛生研究所のサイト)
参考:厚生労働省「ジェネリック医薬品への疑問に答えます ~ジェネリック医薬品Q&A~」 -
ジェネリック医薬品は供給体制に不安がある。安定供給されるのか?
ジェネリック医薬品の安定供給に関しては各製薬企業のみならず、国、業界をあげて取り組んでいます。
解説国民及び医療関係者の皆様が安心してジェネリック医薬品を使用して頂けるよう、行政、医療関係者、医薬品業界など国全体で取り組む施策として平成25(2013)年に「後発医薬品のさらなる使用促進のためのロードマップ」が策定されました。
また令和元(2019)年度、供給不安を予防する目的で日本製薬団体連合会では自己点検チェックリストを作成、加盟団体に配布し、また行政と連携して医薬品供給調整スキームを構築しました。令和2(2020)年度からは厚労省が主催し、有識者らで構成される安定確保策に関する関係者会議が発足し、課題や今後の対応について議論を行っています。
「後発医薬品のさらなる使用促進のためのロードマップ」(日本ジェネリック製薬協会のサイト)
このロードマップにおいては安定供給に関して、業界団体が新たに「ジェネリック医薬品供給ガイドライン」を作成すること、これに準拠した「安定供給マニュアル」を各企業が作成すること、また供給を継続して確保する体制を整備すること等が規定されており、各企業はそのガイドラインに従い安定供給マニュアルを作成し積極的に取り組んでいます。
ジェネリック医薬品の安定供給に関する詳細はこちらをご参照ください。(医療関係者向けジェネリック医薬品についてへのリンク)
日本ジェネリック製薬協会のホームページでは、会員会社の流通の問い合わせ先が確認でき、また供給に何らかの問題が発生したジェネリック医薬品の情報に関しても確認することができます。
流通の問い合わせ先(日本ジェネリック製薬協会のサイト)
製品の供給状況について(日本ジェネリック製薬協会のサイト)
また、平成28年3月からは厚生労働省のホームページ「安定供給体制等を指標とした情報提供項目に関する情報提供ページ」にて、後発医薬品を薬価収載しているすべての製造販売会社の安定供給体制等の情報収集ができるようになりました。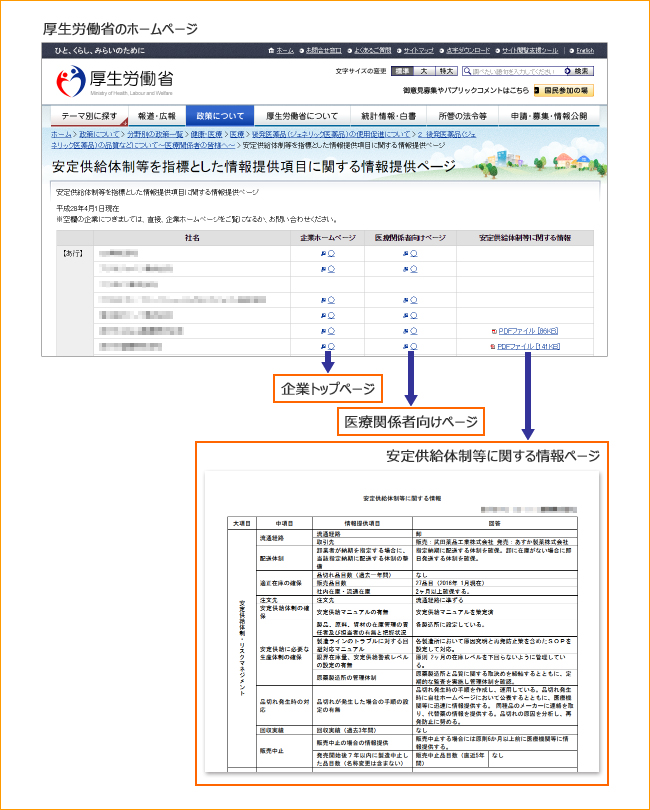
-
ジェネリック医薬品の中には先発医薬品の効能・効果の一部を欠くものがあるが何故か?
先発医薬品の効能・効果、用法・用量の一部に用途特許や再審査期間が付いているために、承認時に先発医薬品の効能・効果、用法・用量の一部が取得できないことがあります。また、先発医薬品が市販後に効能追加等を行ったことにより一時的に異なることがありますが、取得可能となった段階で速やかに申請するよう対応しています。
解説ジェネリック医薬品の効能・効果等が先発医薬品の効能・効果等と一致していない場合があります。これは、先発医薬品の効能・効果等の一部に用途特許や再審査期間が付いている場合や、ジェネリック医薬品の承認取得後に先発医薬品が効能・効果等の追加を行った場合です。いずれの場合にも効能・効果等の一部が欠けているものは、取得可能となった段階で速やかに申請するよう対応しています。
ジェネリック医薬品で先発医薬品と効能・効果等が異なる品目については、当協会のホームページにて公開しています。
効能効果、用法用量等に違いのある後発医薬品リスト(日本ジェネリック製薬協会のサイト) -
ジェネリック医薬品は先発医薬品と比較して情報が少ないと言われているが、どのような試験項目で審査、承認されているのか?
ジェネリック医薬品の申請時には、規格及び試験法、安定性試験、生物学的同等性試験の3つの資料と添付文書記載事項(添付文書案)を提出し審査されます。審査は先発医薬品と同じようにPMDA(医薬品医療機器総合機構)で評価され、先発医薬品と有効性及び安全性が同等であることが確認されなければ承認されません。
解説先発医薬品と比較してジェネリック医薬品の承認申請資料が少なくてすむのは、有効成分に関する有効性・安全性はすでに先発医薬品において確認されているため、同一の有効成分を使用するジェネリック医薬品ではそれらの試験(毒性試験、薬理試験、臨床試験等)を行う必要がないからです。
この考え方は、FDA(アメリカ食品医薬品局)、EMA(欧州医薬品庁)等世界共通です。
ジェネリック医薬品の実施試験が少ないからといって、先発医薬品と比べて有効性、安全性、品質が劣ることは決してありません。
ジェネリック医薬品を製造販売するためには、薬機法(医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律)に基づいて厚生労働大臣から承認を得なければなりません。そのためには、品質、有効性、安全性が先発医薬品と同等で治療学的に同等であることを証明する必要があり、承認申請者は、通常、「規格及び試験方法」、「安定性試験」、「生物学的同等性試験」の3つの資料と添付文書記載事項(添付文書案)を提出することが義務付けられています。
ジェネリック医薬品の申請資料の詳細はこちらをご参照ください。(医療関係者向けジェネリック医薬品についてへのリンク) -
ジェネリック医薬品の添付文書やインタビューフォームに記載されている情報が先発医薬品よりも少ないのは何故ですか、またこれは改善されるのですか?
ジェネリック医薬品は、先発医薬品の長年にわたる臨床使用経験により既にその有効性・安全性が検証されており、健康成人を対象に先発医薬品との生物学的同等性を証明し、品質及び安定性を担保する資料を提出することで、医薬品として承認されます。一方、新薬である新規有効成分の医薬品は、多くの非臨床試験やヒトを対象とした臨床試験等のデータにより有効性・安全性を確認する必要があります。すなわち、ジェネリック医薬品では、必要とされる限られた試験を実施しているため、自ずと添付文書等に記載する情報は少なくなります。ただし、安全性等に関する新しい情報が収集された場合には、先発医薬品と同様、速やかに添付文書を改訂し情報提供することが法的に義務付けられていますので、安全性に関する新しい情報は先発医薬品と同等の情報になっています。
なお、平成31(2019)年より医療用医薬品の添付文書の記載内容が大きく変更になり、ジェネリック医薬品においても新記載要領の添付文書に改訂する際には、先発医薬品と同一・同等の情報を記載して提供することになります。
また、添付文書情報を補完する資料であるインタビューフォームにおいても添付文書と同様に先発医薬品と同一・同等の情報(薬物動態、臨床成績、薬効薬理等)を記載し、情報の充実が図られます。解説先発医薬品の場合は、承認に際しての非臨床・治験データおよび再審査による有効性・安全性のデータが得られているのに対し、ジェネリック医薬品の場合はそれらの先発医薬品のデータを前提として、生物学的同等性試験により同等性を評価していることから、自ずと提供すべき情報は少なくなる傾向にあります。ただし、製造販売後に新たな安全管理情報が得られた場合には、医薬品医療機器等法に基づき、先発医薬品と同様に速やかに添付文書を改訂し情報提供することが義務付けられています。
ジェネリック医薬品の添付文書に記載する情報を充実させるため、平成18(2006)年3月より、生物学的同等性試験データ、溶出試験の適合性、安定性試験データ等を記載することになっています。
医療用医薬品の添付文書については平成31(2019)年4月より、新記載要領に基づく添付文書への改訂が順次行われ、令和6(2024)年3月末までに全ての添付文書が新記載要領に基づくものに改められます。新記載要領に基づく添付文書では、警告・禁忌、重要な基本的注意、相互作用、副作用や適用上の注意などは、原則、先発医薬品とジェネリック医薬品で同一の記載内容になります。
また、薬物動態、臨床成績や薬効薬理については従前どおり、生物学的同等性試験データを記載するとともに、公表されている文献や資料に基づき、先発医薬品と同等の情報提供が求められます。
後発医薬品に係る添付文書の記載に関してはこちらをご参照ください。
「後発品の添付文書等における情報提供の充実について」平成30(2018)年4月13日 薬生薬審発0413第2号、薬生安発0413第1号(厚生労働省のサイト)
「ジェネリック医薬品添付文書記載要領 説明資料について」(日本ジェネリック製薬協会のサイト)また、インタビューフォームについても製品固有の情報(生物学的同等性試験データ、溶出試験の適合性、安定性試験データ等 )の他、新記載要領添付文書の改訂と同様に薬物動態、臨床成績、薬効薬理等の記載の充実を図ります。
更に、調剤・服薬支援については、「医療用医薬品の販売情報提供活動に関するガイドラインに関するQ&Aについて(その3)」において、承認上認められていない用法等である錠剤の粉砕や崩壊・懸濁性及び経管投与チューブの通過性等に関する情報は、インタビューフォームにおいて情報提供することは、医療用医薬品の販売情報提供活動に関するガイドラインにおける医療関係者からの求めがあった場合の情報提供として整理されています。
後発医薬品に係るインタビューフォームの記載に関してはこちらをご参照ください。
「後発医薬品インタビューフォーム作成について」2020 年9 月暫定第1 版(日本ジェネリック製薬協会のサイト) -
ジェネリック医薬品は、先発医薬品に比べてメーカーMRの頻繁な訪問、情報提供が無いのはなぜか?
ジェネリック医薬品は、有効性や安全性の情報が蓄積された上で市場に出るため、医療現場に提供すべき情報は先発医薬品ほど多く求められていません。
しかしながら新たな有効性や安全性の情報を入手した場合には、すみやかな情報提供を行っており、またそれを補完するために、インターネット等を利用した情報提供の充実を図っています。解説先発医薬品は発売された後、それまでの治験段階とは比べものにならないほど多数の臨床使用例が発生するため、治験時にはわからなかった有効性及び安全性等に関する新たな情報が発生します。そこで、比較的高頻度にMRが医療現場を訪問して、担当する医薬品の情報収集と情報提供を活発に行い、医薬品の有効性と安全性の確保に努めることになります。
一方、ジェネリック医薬品は、先発医薬品の使用経験によって有効性、安全性の情報が蓄積された上で市場に出ることになりますので、医療現場に提供すべき情報は、先発医薬品ほど多く求められていません。そのためMR数や訪問頻度は少ない場合が多いと思われます。
それを補完するためにジェネリック医薬品企業は、自社ホームページの充実、インターネットやDMを活用した情報提供等、様々な媒体を活用して情報の提供を行っています。
また業界としても、ジェネリック医薬品の有無や検索、資料請求、各社への問い合わせ等が可能な「ジェネリック医薬品情報提供システム」を運営し、情報提供に取り組んでいます。
「ジェネリック医薬品情報提供システム」(日本ジェネリック製薬協会のサイト) -
生物学的同等性試験の許容域を80%~125%としているが、先発医薬品との治療効果が最大45%の範囲で異なるのか?
生物学的同等性試験で設定されている許容域の幅は、ジェネリック医薬品と先発医薬品の治療効果の差を意味しているわけではありません。この幅は、医薬品を服用した後の血中濃度が、被験者の体質や体調によって大きくばらつく中で、統計的な評価を適確に行うために設定されたものです。この許容域を満たせば、治療効果は安全域をもって同等とみなされています。
実際に承認されている医薬品のデータの検証を実施したところ、先発医薬品とジェネリック医薬品の血中濃度にはほとんど差がありませんでした。解説生物学的同等性試験の許容域は、ジェネリック医薬品と先発医薬品の血中濃度の比の幅を示しているのであって、治療効果そのものの差の幅を示しているわけではありません。通常、医薬品の効果や副作用は有効成分の血中濃度に従って発現しますので、生物学的同等性試験の許容域内であれば、治療効果は同等であると考えられます。
血中濃度に関しては、同じ人が同じ医薬品を服用した場合であっても、体質や体調等が医薬品の吸収、代謝及び排泄に影響を及ぼすなど、除外できない自然のばらつきが常に起こり得ます。生物学的同等性試験の許容域は、このような血中濃度のばらつき等を考慮したうえで、ジェネリック医薬品と先発医薬品の治療効果が同等と評価できる幅を安全域を含めて設定しています(※)。
実際に、PMDA(独立行政法人医薬品医療機器総合機構)が発足した2004年4月1日から2011年1月15日までに承認された経口製剤のジェネリック医薬品について実施された930件の生物学的同等性試験について、ジェネリック医薬品と先発医薬品の平均的な差を比較するための検証を行った結果、それぞれの評価パラメーターの差を先発医薬品に対する比で表すと、Cmaxについては4.6%、AUCtについては3.9%となり、ジェネリック医薬品と先発医薬品の差はほとんどないという結果になりました(図表1参照)。
図表1 ジェネリック医薬品と先発医薬品の差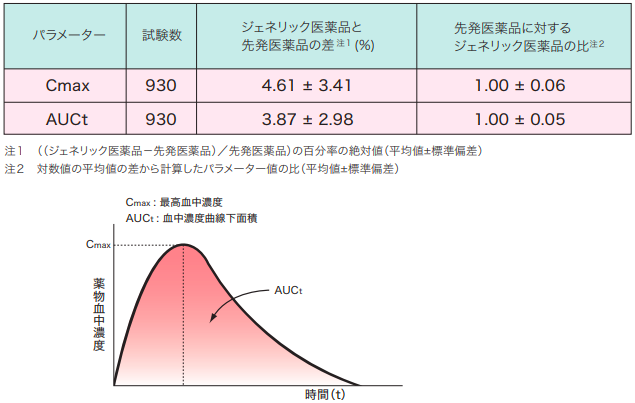
※ 生物学的同等性試験の許容域は、先発医薬品とジェネリック医薬品の比を1と仮定した場合に、先発医薬品とジェネリック医薬品の血中濃度における平均値の比の幅(信頼区間)が100%を中心として±20%(対数変換を行う場合は80%~125%)内にあることを意味しているのであって、ジェネリック医薬品と先発医薬品が最大45%異なり得るということを意味しているわけではありません。
参考:厚生労働省「ジェネリック医薬品への疑問に答えます ~ジェネリック医薬品Q&A~」 -
注射剤については、生物学的同等性試験を実施していないものがあるが、なぜ、同等と言えるのか?
経口剤など体内で吸収された後に血液中に入る製剤と異なり、有効成分が完全に溶解した注射剤で、血管内に直接投与するものについては、血中濃度の推移を変化させる要因がそもそも存在しないため、生物学的同等性試験を行う必要はありません。
解説ジェネリック医薬品の承認にあたっては、基本的には生物学的同等性試験のデータが必要となりますが、有効成分が完全に溶解した注射剤で、血管内に直接投与する医薬品については、生物学的同等性試験の実施は不要と考えます。
通常、医薬品の効果や副作用は、有効成分の血中濃度に従って発現することになります(※)。そのため、先発医薬品と同じ有効成分のジェネリック医薬品は、先発医薬品と同様の血中濃度推移であることが求められます。
しかし、有効成分を均一に溶解させた医薬品を血管内に直接投与する注射剤の場合は、血中濃度の推移を変化させる要因がそもそも存在せず、血管内に投与さえすれば例外なく先発医薬品もジェネリック医薬品も同様の挙動を示すため、血中濃度を測定する必要はありません。含量試験、不純物試験、浸透圧・pHなどの試験及び安定性試験等によって品質が担保されれば、先発医薬品と同等であるということができます。
一方、医薬品を血管外の部位に投与する場合や懸濁剤を血液内等に投与する場合には、有効成分が効果・作用を発揮するためには製剤から放出されて血液内に移行することが必要になりますが、製剤の特性によってその過程は変化する可能性があり、結果として、有効成分の血中濃度がジェネリック医薬品と先発医薬品で異なることがあり得ます。
そのため、血管外に投与するジェネリック医薬品や懸濁剤が先発医薬品と同等であるかどうかを確認するためには、血中濃度が先発医薬品と同様の挙動を示しているかどうかを調べなければならないので、生物学的同等性試験を行う必要があります。
※ 全身作用を期待する医薬品では、有効成分は血液を介して作用発現部位に運ばれ、作用を発現します。血液を介さずに作用発現部位に到達することはありません。
そのため、血液中の有効成分濃度に従って医薬品の効果や副作用は発現すると捉えることができるとされています。なお、TDM(治療薬物モニタリング)は、この考え方に基づき、薬物の血中濃度を測定することで薬効の指標としているところです。
参考:厚生労働省「ジェネリック医薬品への疑問に答えます ~ジェネリック医薬品Q&A~」 -
ジェネリック医薬品で副作用が起こった場合の対応は?
医薬品の副作用情報の収集、提供は、全ての医薬品において、適時適切に実施されることが必要です。したがって、先発医薬品、ジェネリック医薬品の区分に関係なく、GVP制度により副作用情報の収集、提供を行っています。
解説医薬品の安全対策は、先発医薬品であるかジェネリック医薬品であるかにかかわらず、全ての医薬品を対象に、適時適切に実施することが基本です。
平成17(2005)年の改正薬事法施行により、副作用情報の収集、評価・分析、安全確保措置、情報提供といった市販後安全対策の充実・強化のため、製造販売業許可制度が導入され、その許可要件のひとつとしてGVP「医薬品、医薬部外品、化粧品及び医療機器の製造販売後安全管理の基準」が制定され、先発医薬品、ジェネリック医薬品の区別なく、製薬企業に対し市販後安全対策に係る体制整備が求められています。
また医薬品副作用被害救済制度は、先発医薬品、ジェネリック医薬品にかかわらず適用されます(ただし、抗がん剤等一部の医薬品は医薬品副作用被害救済制度の対象外)。
医薬品副作用被害救済制度(医薬品医療機器総合機構のサイト) -
ジェネリック医薬品の販売名はどのようにつけられますか?
現在新たに承認を取得するジェネリック医薬品は、一般的名称を用いた販売名を使用することになっています。またそれ以前に承認されたブランド名の製品に関しても、順次一般的名称を用いた販売名に変更するよう取り組んでいます。
解説平成17(2005)年9月以降、新たに承認申請するジェネリック医薬品の販売名は、「有効成分の一般的名称 + 剤型 + 含量 + 会社名」とするよう統一されています。
日本ジェネリック製薬協会では独自に一般名化の取組みをしてきましたが、平成29年に厚生労働省から発出された「医薬品産業強化総合戦略」に、後発医薬品の一般名化推進が明記され、令和2年3月「後発医薬品使用促進ロードマップに関する調査報告書(厚生労働省医政局経済課 委託事業)」によると、後発医薬品の製造販売承認取得品目数(全品目)のうち、「一般的名称を基本とした販売の品目数」の占める割合は84.0%となっています。
ただし配合剤に関しては、一般名とした場合に製品名が長くなりますので、医薬品の取り違え防止を考慮し、現在は日本ジェネリック医薬品・バイオシミラー学会が中心となって、業界統一のブランド名にするよう勧めています。

