
後発医薬品の生物学的同等性試験ガイドラインの改正のポイントについて
生物学的同等性(BE)試験は、同一有効成分を含有する標準製剤(主に先発医薬品)に対する試験製剤の臨床上の有効性及び安全性の同等性を評価するために実施される試験であり、ジェネリック医薬品が製造販売承認を取得するための重要な試験です。「後発医薬品の生物学的同等性試験ガイドライン等の一部改正について」(薬食審発0229第10号平成24年2月29日)はヒトを対象としたBE評価に関する考え方が示されています。9年前に改正されてから、国際協調、技術開発の向上等、時代の流れに対応したガイドラインの見直しが求められ、令和2年3月19日に「後発医薬品の生物学的同等性試験ガイドライン等の一部改正について」(薬生薬審発0319第1号 令和2年3月19日)が発出されましたので、3つの大きな改正のポイントについて紹介致します。
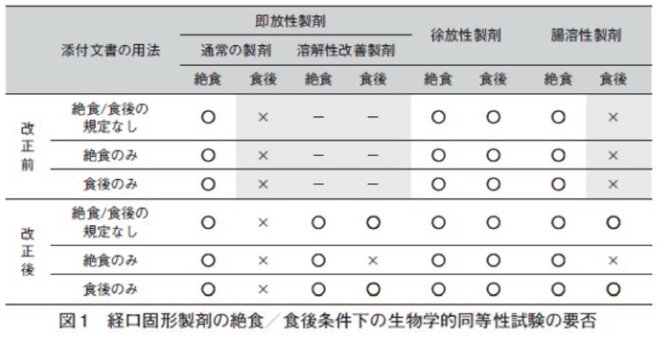
【改正ポイント①】食後条件のBE試験の追加(図1)
BE試験は,製剤間差を評価する感度を上げるために絶食条件下で行われてきました。
通常の即放性製剤においては従来どおり絶食条件下でBE試験を実施しますが、今回の改正でバイオアベイラビリティ(BA)を向上させた溶解性改善製剤については、特殊製剤を食後に服用した場合でも同等に働くかを確認するために絶食条件下のBE試験に加え、食後条件下でも試験を行うことになりました。主な溶解性改善技術として、固体分散体、アモルファス、マイクロエマルジョン、ナノ粒子が改正ガイドラインのQ&Aに記載されています。なお、用法が食前投与のみの製剤は、絶食条件下のBE試験のみで十分とのことです。
また、放出調節製剤の一つである徐放性製剤は以前から食後条件下でのBE試験が求められていましたが、腸溶性製剤についても食後投与時に胃から腸への移動速度が吸収に影響して血中濃度-時間曲線が変化する可能性があるため、絶食下に加え、食後条件下のBE試験も求めることに改正されました。なお、腸溶性製剤についても、用法が食前投与のみの製剤は、絶食条件下のBE試験のみで十分とのことです。
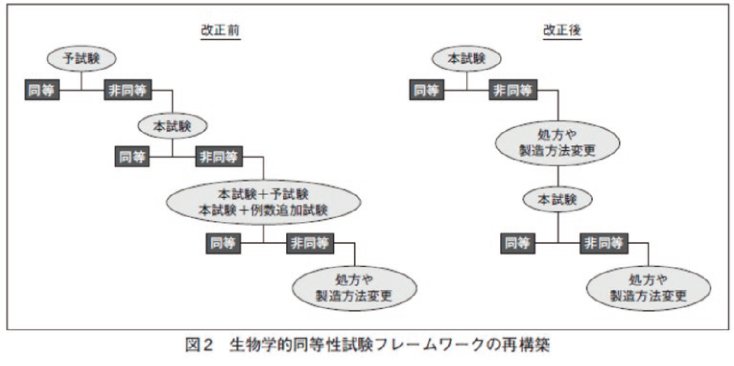
【改正ポイント②】予試験及び例数追加試験の再検討(図 2)
BE ガイドライン改正前は、BE 試験は予試験、本試験及び例数追加試験の 3 段階で評価可能でした。今回の BE ガイドラインの改正では、BE 評価の再構築を行い、予試験や本試験の目的を明確にし、例数追加試験は原則認めないことに変更され,本試験が後発医薬品の開発において検証試験であることが明確になりました。予試験は、本試験の必要例数及び体液採取間隔を含む適切な試験デザインを立案するための情報を得ることを目的とします。BE の同等・非同等の評価は本試験のみで行い.非同等の場合には、製剤処方の変更や製造方法の変更を行うことになりました。なお、予試験実施の有無に関わらず、申請時には本試験の試験計画の妥当性について十分説明できるようにしておく必要があります。例数追加試験が原則認められなくなったことから、本試験では被験者の脱落例も考慮した上で、十分な例数で試験を行うことになります。ただし、予試験による情報を含む事前情報から個体内分散 ( 個体間のバラつき ) が大きく、BE を示すことができない可能性がある場合は中間解析を行うことが可能です。この場合は、プロトコル立案時から中間解析を計画し、第一種の過誤確率を適切な方法によって制御することが必要となります。
【改正ポイント③】外国人を対象とした BE 試験の導入と国内標準製剤使用の明確化
BE ガイドライン改正前は、外国人を対象に実施された BE 試験データは原則受け入れられませんでした。しかし、近年では、海外で実施された臨床試験が適切に実施されていたかの確認が行える体制整備がなされたため、海外で外国人を対象に実施された BE 試験データも受入れられることになりました。
次に、人種差を考慮する必要性について検討し、一般的な BE 試験は標準製剤と試験製剤間の相対比較であるため、被験者の民族的差異が試験結果に与える影響として無視できる程度に小さい場合に限り、海外で外国人を対象に実施された BE 試験は受入れ可能としました。しかし、標準製剤と試験製剤の溶出率の間に「著しい差」がある場合や、製剤特性により胃液酸度をはじめとする消化管の生理学的要因の民族的差異が BE の評価に影響すると考えられる場合には、日本人を対象とした BE 試験を実施する必要があります。
また、BE 試験は日本における後発医薬品の製造販売承認を取得するための試験であるため、日本で承認され流通している製剤を標準製剤として製剤間差を比較したデータが必要であることが明確となりました。
今回の BE ガイドラインの改正の最大のポイントは、本試験の位置付けが明確に規定され、本試験以外で BE の評価が認められなくなった点です。BE 評価をする上で、予試験や事前情報で例数の設計や測定の時間ポイントの妥当性等、BE 試験デザインの妥当性をより慎重に判断する必要があり、より科学的及び合理的に BE 評価が行われる体制が整備されました。
〈参考文献〉
1. 厚生労働省,後発医薬品の生物学的同等性試験ガイドライン(薬生薬審発 0319 第 1 号 別紙 1)
https://www.pmda.go.jp/files/000234565.pdf
2. 厚生労働省,後発医薬品の生物学的同等性試験ガイドライン Q&A(事務連絡 別紙 1)
https://www.pmda.go.jp/files/000234569.pdf
3. 「後発医薬品の生物学的同等性試験ガイドラインの近代化と再構築」独立行政法人 医薬品医療機器総合機構
ジェネリック医薬品等審査部 栗林 亮佑(後発医薬品品質情報、令和2年8月、No.14、P5-9) ※ 図1-2 は本資料からの抜粋