
ジェネリック医薬品について
ジェネリック医薬品とは
ジェネリック医薬品(後発医薬品)とは、先発医薬品と同等であることが認められた、先発医薬品に比べて低価格な医薬品です。欧米では有効成分の一般名(generic name)で処方されるため、「ジェネリック」という言葉で呼ばれています。ジェネリック医薬品は、先発医薬品と同じ有効成分を含有し、先発医薬品と同一の効能・効果を有します(一部の効能・効果の場合もあり)。ジェネリック医薬品は、患者さんから得られた先発医薬品の有効性や安全性等の社会的財産を無償で使用して社会に還元するものであることから、低コストで提供することができます。
再審査制度
ジェネリック医薬品は、先発医薬品の再審査期間が終了した後に申請することができます。
再審査制度とは、先発医薬品の承認後に有効性と安全性を再度確認する制度です。治験ではわからない副作用や医療現場での使われ方による影響を調べるため、先発医薬品の承認後の一定期間、先発医薬品企業が医療機関で使用されたデータを集め、確認します。この先発医薬品の承認後の有効性と安全性の確認期間を再審査期間と言います。再審査によって、実際の医療現場における先発医薬品の有効性と安全性の確認が終了した後に、ジェネリック医薬品の申請を受け付けることになっています。
新有効成分の医薬品の再審査期間は、製造販売承認の日から8年間です。小児での安全性を確認する臨床試験を行った場合には、さらに2年間、再審査期間が延長されます。ジェネリック医薬品は、申請から承認までに1年間かかりますので、新有効成分を含有する医薬品の承認の日から9年以上の間、そのジェネリック医薬品は承認されません。
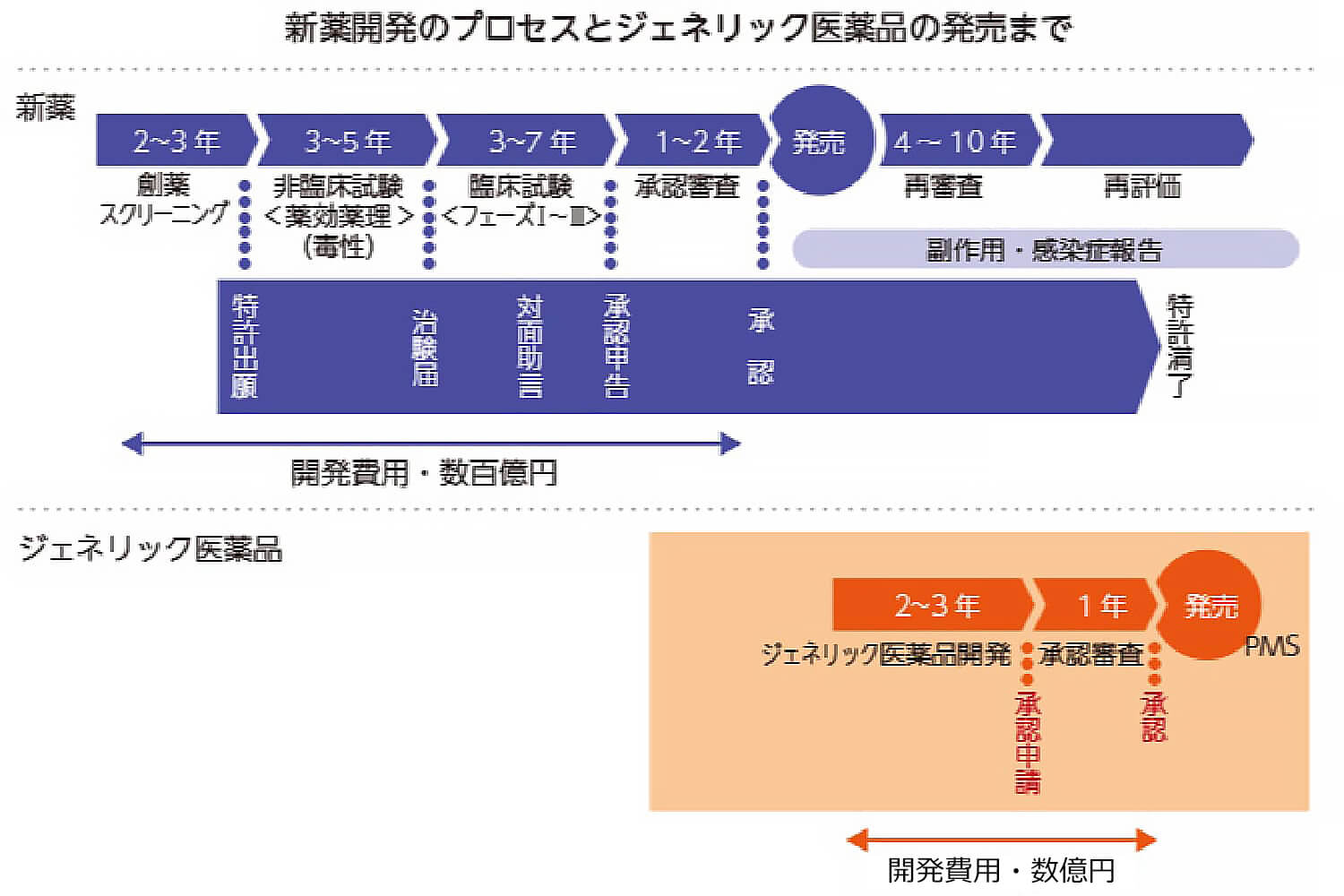
※上図はイメージです。実際の開発期間や特許期間については、様々なパターンがあります。
ジェネリック医薬品に関係する特許について
知的財産権
医薬品産業においても、他の産業と同様に他社の知的財産権を侵害しないように、十分な調査と検討を行います。
知的財産権には、音楽、映画、絵画などの著作物を保護する著作権、発明を保護する特許権、考案を保護する実用新案権、デザインを保護する意匠権、商品やサービスなどを区別するためのマークに化体した信用を保護する商標権などがあります。医薬品産業においては、特に、特許制度が重要です。
特許制度とは、発明を公開した者に対し、一定の期間その利用についての独占的な権利を付与することによって発明を奨励するとともに、第三者に対しても、この公開された発明を利用する機会を与え、もって産業の発達に寄与しようとするものです。そのため、特許権の存続期間が終了した後は、何人でも自由にその発明を利用することができ、それによって社会一般が広く益されるようになります。特許制度はあらゆる産業を対象として、その発達を目的とした制度です。医薬品産業においては、先発医薬品企業は、物質特許が存続する期間中は他社の参入ができないことによる独占的な利益を得ることができ、それにより、開発コストを回収することが可能になります。本来、公正な競争こそが市場経済の命題ですが、特許制度は、公正な競争の例外として、出願の日から20年(医薬品については、さらに最長5年間延長可能)の独占権を付与することにより、発明の公開を促しています。公正な競争の例外ですから、独占期間が不当に長いと、公正な競争が損なわれてしまい、却って産業の発達の妨げとなります。医薬品においては、患者さんが安価なジェネリック医薬品を選択する自由を妨げることになります。
医薬品関連特許は、権利範囲を限定する要素によって、慣習的に呼び分けられています。例えば、物質の構造のみによって権利範囲を限定する「物質特許」、物質の製造方法で権利範囲を限定する「製法特許」、使用用途や対象疾患によって権利範囲を限定する「用途特許」、使用方法や使用量によって権利範囲を限定する「用法用量特許」、製剤技術によって権利範囲を限定する「製剤特許」、複数の有効成分によって権利範囲を限定する「配合剤特許」や「併用特許」、物質の結晶型によって権利範囲を限定する「結晶特許」などがあります。
これらの慣習的に呼び分けられる各種特許の夫々の例を下表に示します。
権利範囲の限定方法による慣習的な呼び方ですので、現実には、複数種類の限定が組み合わされ、例えば、用途特許でもあり製剤特許でもあるといったことも多く、必ずしも、一意に、呼び分けることができるわけではありません。
医薬品関連特許の例
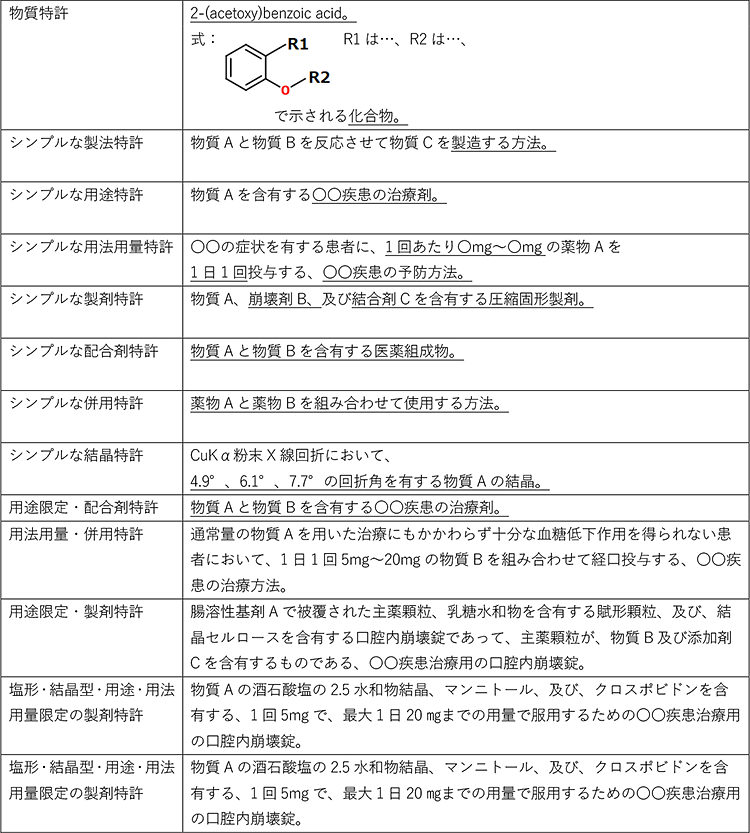
通常、「先発医薬品の特許期間の満了」とは「物質特許の満了」を指します。製法特許や製剤特許の特許期間が残っている場合は、それらの特許を回避して製造します。
法的基準について
医薬品に関する基準
医薬品は、薬機法※のもと、下記のようなさまざまな規制を遵守して開発・製造販売されていますが、その規制は先発医薬品独自のものでなく、ジェネリック医薬品も先発医薬品と同じものとなっています。
※薬機法:「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」
日本における医薬品の法的基準
| GQP(Good Quality Practice) | 医薬品、医薬部外品、化粧品及び医療機器及び再生医療等製品の品質管理の基準 |
|---|---|
| GLP(Good Laboratory Practice) | 医薬品の安全性に関する非臨床試験の実施の基準 |
| GCP(Good Clinical Practice) | 医薬品の臨床試験の実施の基準 |
| GMP(Good Manufacturing Practice) | 医薬品及び医薬部外品の製造管理及び品質管理の基準 |
| GVP(Good Vigilance Practice) | 医薬品、医薬部外品、化粧品、医療機器及び再生医療等製品の製造販売後安全管理の基準 |
| GPSP(Good Post-marketing Study Practice) | 医薬品の製造販売後の調査及び試験の実施の基準 |
ジェネリック医薬品の承認申請
ジェネリック医薬品の承認申請は通常、「規格及び試験方法」、「安定性試験」、「生物学的同等性試験」の3つの資料と添付文書記載事項(添付文書案)の提出によって審査されます。
医薬品の申請資料
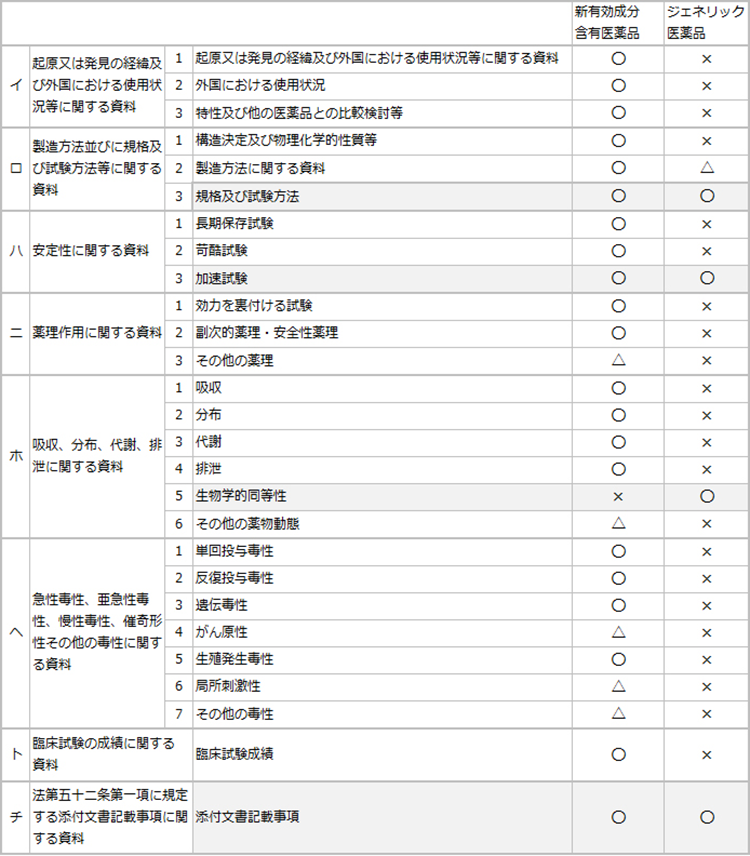
○:添付×:添付不要 △:個々の医薬品により判断される
(1)規格及び試験方法
原薬及び製剤の規格及び試験方法(性状、確認試験、純度試験、溶出試験、含量試験等)における基準は、先発医薬品、ジェネリック医薬品において同様であり、国の厳格な審査を受けて承認されます。また、ジェネリック医薬品の承認審査においては、原薬と製剤両方に対して、対照となる先発医薬品と同等またはそれ以上の規格設定が承認の条件となっています。
(2)安定性試験
医薬品の市場流通期間中の品質を保証するため、安定性試験を実施しています。承認申請時に実施する加速試験(通常40℃±2℃、75%RH±5%RH、6ヶ月)の他、長期保存試験(通常25℃±2℃、60%RH±5%RH、3年間)も実施し、有効期間内の品質に問題の無いことが確認されています。また、医療機関での使用時の品質を保証するために無包装状態下における安定性試験等を実施し、使用状況下での情報が提供されています。
(3)生物学的同等性試験
ジェネリック医薬品は対照となる先発医薬品と生物学的に同等であることをもって承認されます。生物学的同等性が実証できれば、改めて臨床試験を行うことなく、両医薬品の臨床における有効性と安全性の同等性が担保されます。
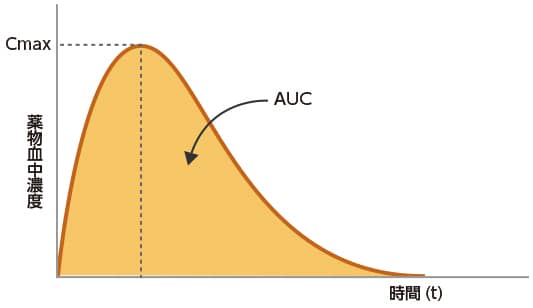
生物学的に同等とは、ジェネリック医薬品と先発医薬品のバイオアベイラビリティ(生物学的利用能:薬物が血管外投与部位から全身循環血中に入る速度と量)が同等であることをいい、ジェネリック医薬品と先発医薬品を個々に投与したときの、最高血中濃度(Cmax)、血中濃度時間曲線下面積(AUC)等を比較することにより、統計的に同等であるか否かを評価します。
生物学的同等性の評価方法は「後発医薬品の生物学的同等性試験ガイドライン」に定められており、ガイドラインに定められた試験方法や基準は最新の科学的知見に基づく世界標準の考え方です。
生物学的同等性の試験方法
ジェネリック医薬品(試験製剤)と先発医薬品(標準製剤)を用いて、原則としてクロスオーバー法で、健康成人を対象に臨床常用量を投与し、その薬物(有効成分)の血中濃度の時間推移を測定します。
クロスオーバー法とは
クロスオーバー法とは、同一被験者が一定期間を空け、先発医薬品とジェネリック医薬品をそれぞれ交互に服用する試験方法です。
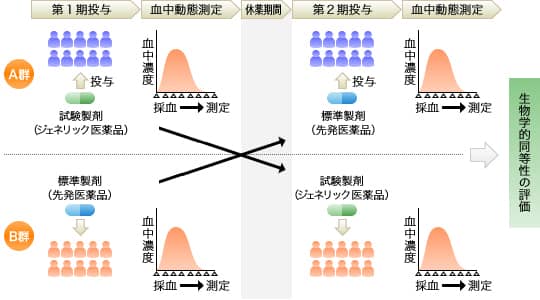
生物学的同等性の評価方法
生物学的同等性試験の評価は、試験によって得られたデータを統計的に解析して行います。CmaxとAUCを主なパラメータとし、最高血中濃度到達時間(tmax)や血中濃度半減期(t1/2)などは参考パラメータとして用います。
作用発現時間の差が臨床的有用性に影響を与える可能性がある場合にはtmaxもCmax、AUCとともに生物学的同等性試験の判定パラメータとなることがあります。
有効成分の血中動態の測定
AUC:血中薬物濃度一時間曲線下面積(area under the blood concentration time curve)
生体内に投与された薬物の血中濃度を経時的に表したグラフの曲線と時間軸によって囲まれた部分の面積。薬物のバイオアベイラビリティやクリアランスの指標となる。
t max:最高血中濃度到達時間(maximum drug concentration time)
生体内に投与された薬物が最高血中濃度(Cmax)に達するまでの時間。
t 1/2:血中濃度半減期
生体内に投与された薬物の血中濃度が半減するまでの時間。
ジェネリック医薬品の品質確保について
先発医薬品、ジェネリック医薬品を含むすべての医薬品は、同じ基準で管理、製造されています。
(1)品質保証の基準(GQP)
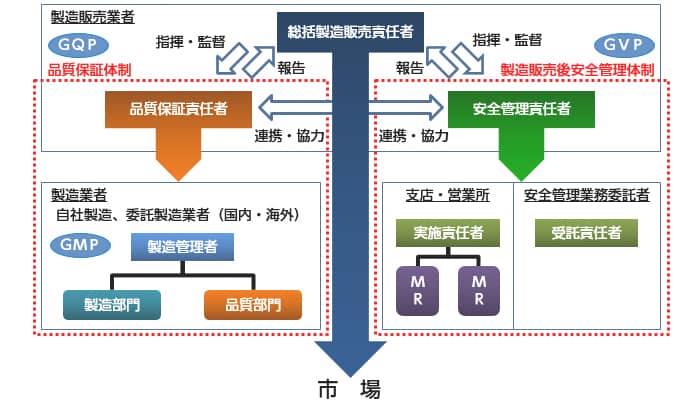
平成17(2005)年4月の改正薬事法の施行により、使用する原薬から包装された最終製品に至る医薬品の全ての製造(自社を含む国内外の工場にて行われた製造行為すべて)は、医薬品製造業の許可(責任)のもとで行われることになりました。これらを総合的に管理し、製造販売する医薬品の品質を保証するのが製造販売業者であり、製造販売業者が守るべき基準として「医薬品、医薬部外品、化粧品及び医療機器の品質管理の基準(GQP)」が制定されています。
(2)品質保証体制
製造販売業者は総括製造販売責任者の監督のもと、GQPに基づき品質保証責任者が製造販売する医薬品の品質保証にあたっています。原薬から最終製品に至るまでの医薬品の製造に関わる全ての製造所を総合的に管理し、出荷される医薬品の品質を確保します。ジェネリック医薬品の製造所において、ジェネリック医薬品だけが製造されているのではなく、多くの先発医薬品も受託製造されており、先発医薬品、ジェネリック医薬品を問わずこれら製造所の管理水準若しくは状態がその品質を決定します。
また、製造販売業者は行政によりGQPを遵守した品質保証を行っているか定期的な査察を受けることも義務付けられています。
医薬品製造販売業の管理体制
(3) 製造管理及び品質管理の基準 (GMP)
製造所における医薬品の製造は「医薬品及び医薬部外品の製造管理及び品質管理の基準(GMP)」及び「薬局等構造設備規則」に基づき、ソフトとハードの両面から適切に管理されています。各製造所は製造する医薬品の種類に応じた適切なクリーン環境を整えており、その製造は教育訓練を受けた作業者により、定められた手順に基づき行われています。また、品質部門により、承認内容を遵守した製造方法、変更管理等の品質マネジメントへの取組み及び基本となる原薬、製造の各工程、中間製品並びに最終製品の品質は厳重に試験・検査されています。更に行政による定期的な査察を受けることも義務付けられています。
(4)後発医薬品品質確保対策事業における品質試験・評価
厚生労働省では、後発医薬品の使用推進の観点から、平成19(2007)年10月に「後発医薬品の安心使用促進アクションプログラム」、平成25(2013)年4月に「後発医薬品のさらなる使用促進のためのロードマップ」をそれぞれ策定し、①安定供給等、②品質確保、③ジェネリック医薬品メーカーによる情報提供、④使用促進に係る環境整備及び⑤医療保険制度上の事項に関し、使用促進策に係る目標を設定し、国及び関係者が取組むべき施策を明らかにしてきました。
その中で品質確保の観点から、厚生労働省では平成20(2008)年度より「後発医薬品品質確保対策事業」として、市場に流通するジェネリック医薬品を対象に溶出試験等の品質検査を実施し、その検査結果を積極的に公表しています。
本事業としての品質試験は、市場に流通しているジェネリック医薬品を対象に、年度毎に計画的に実施されています。
(5)ジェネリック医薬品品質情報検討会
ジェネリック医薬品の品質に対する信頼性の確保の為ため、厚生労働省の委託を受けて平成20(2008)年、国立医薬品食品衛生研究所に、「 ジェネリック医薬品品質情報検討会 」が設置されました。本検討会では有識者の協力を得て、ジェネリック医薬品の品質に関する情報について学術的観点から検討するとともに、前述の後発医薬品品質確保対策事業における必要な試験・評価を実施し、検討結果を国立衛生研究所のホームページにて公表しています。
http://www.nihs.go.jp/drug/ecqaged/kentou-shingi.html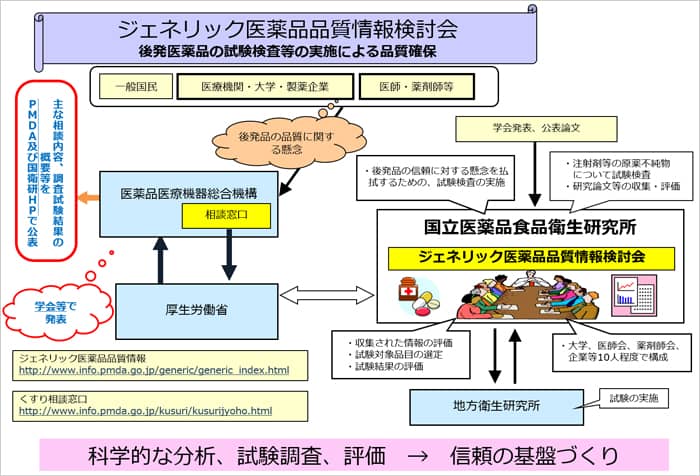
また、ジェネリック医薬品品質情報検討会での検討結果等を踏まえて、厚生労働省から「医療用医薬品最新品質情報集(ブルーブック)」が発刊されています(有効成分を順次追加)。
これは品質再評価において実施されたジェネリック医薬品の溶出試験結果を記載した日本版オレンジブックを改良・発展させたもので、ジェネリック医薬品の品質確認検査及び品質に関する情報を有効成分ごとに体系的にまとめたデータシートであり、溶出試験結果に加え、生物学的同等性試験結果や物性データも掲載しています。
ブルーブックは、国立医薬品食品衛生研究所のホームページでも公開されています。
(6)後発医薬品品質情報
更に、医療関係者や一般の方に対し、ジェネリック医薬品品質情報検討会の情報をはじめ、ジェネリック医薬品の品質に関する情報を、より積極的に発信するために、厚生労働省から平成26(2014)年4月より「 後発医薬品品質情報 」が発刊されています。
「 後発医薬品品質情報 」の電子版は厚生労働省のホームページで公開されています。
https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iyakuhin/kouhatsu_iyakuhin/また、医療関係者や一般の方に対し、ジェネリック医薬品の使用に際し有用な情報を提供することを目的に、厚生労働省から平成26年4月より「 後発医薬品品質情報 」が発行されています。 「 後発医薬品品質情報 」にはジェネリック医薬品品質情報検討会が検証しているジェネリック医薬品の品質に関する情報等が掲載されています。

- 後発医薬品品質情報No.17 (2023年10月31日掲載)
- 後発医薬品品質情報No.16 (2023年2月28日掲載)
- 後発医薬品品質情報No.15 (2022年9月29日掲載)
- 後発医薬品品質情報No.14 (2020年8月7日掲載)
- 後発医薬品品質情報No.13 (2020年3月27日掲載)
- 後発医薬品品質情報No.12 (2019年9月13日掲載)
- 後発医薬品品質情報No.11 (2019年1月18日掲載)
- 後発医薬品品質情報No.10 (2018年7月20日掲載)
- 後発医薬品品質情報No.9 (2018年1月26日掲載)
- 後発医薬品品質情報No.8 (2017年5月31日掲載)
- 後発医薬品品質情報No.7 (2016年12月22日掲載)
- 後発医薬品品質情報No.6 (2016年06月30日掲載)
- 後発医薬品品質情報No.5 (2016年02月12日掲載)
- 後発医薬品品質情報No.4 (2015年11月27日掲載)
- 後発医薬品品質情報No.3 (2015年05月28日掲載)
- 後発医薬品品質情報No.2 (2014年12月22日掲載)
- 後発医薬品品質情報No.1 (2014年04月24日掲載)
ジェネリック医薬品の安全管理について
(1) 安全管理情報の収集・評価・提供
先発医薬品と同様に、ジェネリック医薬品を適正に使用していただくために、製造販売後における安全管理基準等を遵守し、副作用情報等の迅速かつ適正な収集・評価・提供を行っています。また、医療関係者の他、患者・一般の方からのお問い合わせに対応できるよう、各企業ではくすり相談窓口を設置しています。
(2) 製造販売後安全管理
平成17(2005)年4月の改正薬事法の施行により、製造販売後の安全対策を強化する目的で製造販売業の許可要件として、総括製造販売責任者、安全管理責任者等の設置が義務付けられました。
ジェネリック医薬品企業においても、先発医薬品企業と同様に製造販売後の安全管理を適正かつ円滑に運用するための体制を整えています。
(3)製造販売後調査
ジェネリック医薬品は先発医薬品の再審査により既に有効性と安全性が検証されているため、先発医薬品で義務づけられている製造販売承認後の使用成績調査は通常行われませんが、成分によっては更に特定使用成績調査を求められることがあります。これらの調査は先発医薬品と同様に医薬品管理リスク計画を提出し、医療機関での使用実態を安全性を中心に調査収集、評価・解析を行い、安全性について評価した結果を医療機関にフィードバック(伝達)することで安全対策を図っています。
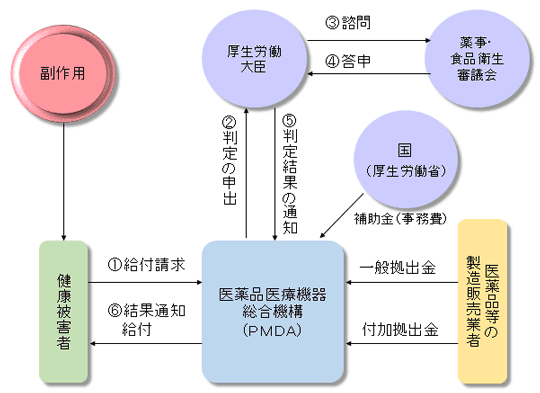
(4)医薬品副作用被害救済制度
「 医薬品副作用被害救済制度
」は、医薬品等を適正に使用したにもかかわらず発生した副作用により健康被害を受けた方に対して、医療費等の給付を行い、被害を受けた方の迅速な救済を図ることを目的として、昭和55(1980)年に創設された医薬品医療機器総合機構法に基づく公的な制度です。
「 医薬品副作用被害救済制度
」は医療用医薬品(先発医薬品、ジェネリック医薬品)、一般用医薬品を問わず発生した健康被害に対して、医療費の給付等の救済を受けることができます(ただし救済には審査があり、また抗がん剤等一部対象外の医薬品もあります)。
医療費等の給付に必要な費用は、先発医薬品、ジェネリック医薬品とわず、全ての医薬品製造販売業者等からの拠出金で賄われています。
ジェネリック医薬品の安定供給について
ジェネリック医薬品の製造販売業者は、先発医薬品の製造販売業者と同様に、医療用医薬品を扱うものの使命として、良質なジェネリック医薬品を医療機関・調剤薬局に安定供給するべく鋭意努力しています。
具体的には、国民及び医療関係者の皆様が安心してジェネリック医薬品を使用して頂けるよう、行政、医療関係者、医薬品業界など国全体で取り組む施策として平成25(2013)年に策定された「後発医薬品のさらなる使用促進のためのロードマップ」に則り、納品までの時間短縮、供給ガイドラインの作成、安定供給マニュアルの作成、業界団体による支援、製造所に対する品質管理の徹底、品切れを起こした場合の迅速な対応、原薬調達や供給能力などに関する計画の作成について真摯に取り組んでいます。
ジェネリック医薬品供給ガイドラインと安定供給マニュアル
平成25(2013) 年に策定された「 後発医薬品のさらなる使用促進のためのロードマップ」において、ジェネリック医薬品の製造販売業者が「安定供給マニュアル」を作成するための指針となる「 ジェネリック医薬品供給ガイドライン 」を業界団体で策定することとされました。 このガイドラインでは、これまでに経験した品切れ事例を調査・分析し、その原因を排除するために立てられた対策を参考に、各企業が「安定供給マニュアル」を作成するにあたって留意すべきことを、12の手順として定めております。
<ジェネリック医薬品供給ガイドラインに記載されている手順>
- 原薬の安定確保に関する手順
- 在庫管理に関する手順
- 生産管理に関する手順
- 他社に製剤を製造委託する場合の手順
- 配送に関する手順
- 安定供給に関連する情報の収集、評価に関する手順
- 安定供給に支障をきたすおそれがある案件発生時の対応に関する手順
- 品切れ等発生時の対応に関する手順
- 供給停止に関する手順
- 記録に関する手順
- 自己点検に関する手順
- 制定改廃に関する手順
すべてのジェネリック医薬品の製造販売業者は、本ガイドラインに準拠した「安定供給マニュアル」を作成・運用し、適切な需要予測に基づく原薬等の確保、製造管理・品質管理の徹底により、医薬品製造販売業者として安定供給の責任を果たすことに努力しています。
また、近年、過去の供給問題とは異なる事案による安定供給に問題が生じるケースがあることから、令和2(2020)年2月、日本製薬団体連合会(日薬連)はCOVID-19感染拡大によるサプライチェーンの寸断などで発生する出荷停止事案に対応するため、「新型コロナウイルスに関連した感染症発生に関する事務連絡への対応と医療用医薬品供給調整スキームの策定について」を発出し、医薬品の安定供給に支障を来す事態を早期に把握し、企業が相互に協力できるスキームを策定し、代替薬をリスト化するなどの取り組みを行っています。
供給不安等が生じた場合に医療機関等への影響を最小限に留め、情報の早期連絡と迅速な対応体制を構築するため、『「医薬品供給調整スキーム」における医療用医薬品の供給不安発生時の供給調整に関する手順』について、当面の間、以下のように改めて運用することとしました。
- 各社の連絡窓口を予め日薬連に登録しておくこと。
- 自社製造販売品目で本スキームの対象となる製品について、予め当該医薬品の同一成分薬、及び代替薬の製品名及びメーカーをリスト化しておくこと。
- 供給不安が見込まれる場合、可能な限り早期(2箇月前を想定)に、厚生労働省医政局経済課に報告すること。
当協会会員会社における製品の供給状況については 「 製品の供給状況について 」 をご参照ください。
また、会員各社の取り扱い製品および取り扱い販売業者等の詳細については、「 流通に関する問い合わせ
」から各社へお問い合わせください。
ジェネリック医薬品の製剤工夫と添加剤について
(1)ジェネリック医薬品と製剤工夫
ジェネリック医薬品は、先発医薬品と全く「同じ」である必要はなく、服用性や医療関係者・患者さんの利便性等を考慮した製品もあります。
ジェネリック医薬品の製剤工夫例
<服用性への工夫>
- 口腔内崩壊錠
- 苦味マスキング技術
- 新剤型・新規格追加
<医療関係者・患者さんの利便性の考慮>
- 錠剤への製品名のカタカナ印刷
- PTPシートなどの表示(ピッチコントロール、薬効記載等)
- 保存条件の改善(冷所保存→室温保存等)
- 切り取りタグ付き個装箱
<安全性の工夫>
- ガラスアンプルからポリボトルやプレフィルドシリンジ
- 抗がん剤調整時の被爆防止のためのシュリンク包装等
(2)添加剤について
ジェネリック医薬品と先発医薬品では、使用している添加剤が異なる場合があります。例えば先発医薬品が製剤特許を有している場合などは、ジェネリック医薬品は先発医薬品と異なる添加剤を使用することがあります。その他、製剤工夫のために添加剤を変えたりすることもあります。
医薬品に使用する添加剤は先発医薬品、ジェネリック医薬品ともに、その製剤の投与量において薬理作用を示したり、有効成分の治療効果を妨げたりするものは使用できません(日本薬局方製剤総則)。使用前例があり、安全性が確認されている添加剤が使用されています。添加剤が異なっても、有効性や安全性に影響はありません。
ただし、アレルギーをお持ちの患者さんは、先発医薬品、ジェネリック医薬品を問わず、添加剤の中でアレルギーを起こすものがあるかもしれませんので、添付文書には原則全ての添加剤が記載されています。
ジェネリック医薬品と添加剤についての詳細は 「医薬品添加剤について」(PDF 496 kb)
をご参照ください。
ジェネリック医薬品を取巻く環境について
国民医療費の伸び、患者の負担増
高齢化や医療技術の進歩などによって国民医療費は年々増加していますが、一方で経済環境や財政環境は厳しい状況にあり、医療制度改革は大きな課題となっています。医療の質を落とすことなく医療の効率化を図り、国民皆保険制度を維持していくために、医療資源の効率化を通じて国民医療費の適正化を図ることが求められています。
我が国の国民医療費は平成30(2018)年度に国民医療費は43兆を超え、そのうち約2割以上を薬剤費が占めています。ジェネリック医薬品の有効利用により薬剤費の適正化が可能です。
国民医療費・対国内総生産及び対国民所得比率の年次推移
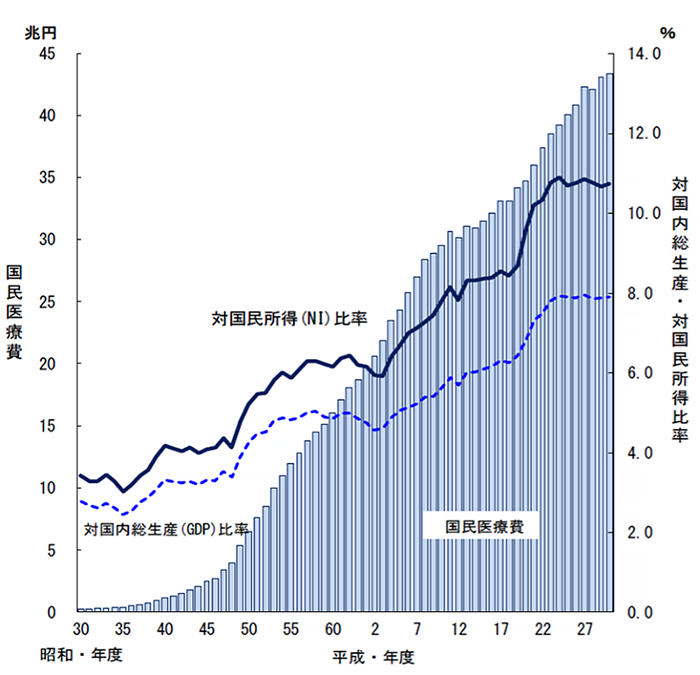
平成30年度 国民医療費の概況(厚生労働省)
ジェネリック医薬品使用促進の動き
平成14(2002)年4月以降、ジェネリック医薬品の使用促進につながる診療報酬・調剤報酬改定や処方せん様式の変更などが行われてきていますが、ジェネリック医薬品の使用促進に関して政府方針としての位置づけが具体的目標をもって明確化されたのは平成19(2007)年です。
平成19(2007)年6月
経済財政改革の基本方針2007において「平成24(2012)年度までにすべての医薬品に対するジェネリック医薬品のシェア(数量ベース)を30%以上に」という数値目標を策定しました。
平成19(2007)年10月
厚生労働省はジェネリック医薬品に関する安定供給・品質確保・情報提供等の充実・向上と使用促進を図るための「後発医薬品の安心使用促進アクションプログラム」を策定しました。
平成20(2008)年度診療報酬・調剤報酬改定
- 「後発医薬品調剤体制加算」:保険薬局のジェネリック医薬品調剤率(処方せん枚数)30%以上に加算。
- 「処方せん様式の変更」:「変更可能の場合に署名」から「変更不可の場合に署名」に変更。
- 「保険薬局及び保険薬剤師療養担当規則」の改正:保険薬局や保険薬剤師にジェネリック医薬品の調剤に必要な体制確保や患者さんに対する説明義務などを新しく規定。
平成22(2010)年度診療報酬、調剤報酬改定
- 後発医薬品調剤体制加算の段階的評価:処方せん枚数割合から後発医薬品の数量ベース割合に変更、段階的に点数を設定(20%以上6点、25%以上13点、30%以上 17点)。
- 医療機関の後発医薬品使用体制加算を新設(採用割合20%以上)。
- 含量違い、類似別剤型への変更調剤(10mg1錠 → 5mg2錠、カプセル→錠)
- 「保険医療機関及び保険医療養担当規則等」の改正:保険医である医師や歯科医師に患者さんがジェネリック医薬品を選択しやすいように努めることを規定。
平成24(2012)年度診療報酬、調剤報酬改定
- 後発医薬品調剤体制加算の段階的評価の見直し(22%以上5点、30%以上15点、35%以上19点)
- 薬剤情報提供文書を活用した後発医薬品の情報提供への評価
- 医療機関の後発医薬品使用体制加算の見直し(20%以上28点、30%以上35点)
- 一般名処方の推進
- 処方せん様式の変更(薬剤ごとに変更可否を明示)
平成25(2013)年4月
「後発医薬品のさらなる使用促進のためのロードマップ」を策定
厚生労働省はジェネリック医薬品のシェアを平成30年度までに60%以上(置き換え可能な市場における割合)にすることを目標として「後発医薬品のさらなる使用促進のためのロードマップ」を策定しました。このロードマップには(1)さらなる使用促進の必要性(2)新たな目標の設定とモニタリングの強化(3)国、企業、保険者等の具体的な取り組みおよび課題等について明記されています。
平成26(2014)年度診療報酬、調剤報酬改定
- 後発医薬品調剤体制加算の段階的評価の見直し(ロードマップで示された新指標に基づく計算方式 55%以上18点、65%以上22点)
- 調剤薬局において一般名処方せんの際に後発医薬品を調剤しなかった場合は、診療報酬明細書の摘要欄にその理由を記載しなければならないことを規定
- DPC病院の機能評価係係数2に後発医薬品指数を新設
平成27(2015)年6月
平成27年6月30日、「経済財政運営と改革の基本方針2015」(骨太方針)が経済財政諮問会議での答申を経て、閣議決定されました。この中でジェネリック医薬品のシェアを「2017年(平成29年)央に70%以上とするとともに、2018年度(平成30年度)から2020年度(平成32年度)末までの間のなるべく早い時期に80%以上とする。」とされています。
平成28(2016)年4月
平成28(2016)年度診療報酬、調剤報酬改定
- 後発医薬品調剤体制加算の段階的評価の見直し(65%以上18点、75%以上22点)
- 後発医薬品使用体制加算の段階的評価の見直し(新指標に基づく計算方式に変更、70%以上42点、60%以上35点、50%以上28点)
- 院内処方を行う診療所で後発医薬品を推進している場合の評価として、外来後発医薬品 使用体制加算を新設(70%以上4点、60%以上3点)
- DPC病院の機能評価係数2の後発医薬品指数の上限を引き上げ(60%→70%)
- 一般名処方のさらなる推進(後発医薬品が存在する全ての医薬品が一般名処方されている場合3点、1品目でも一般名処方された医薬品が含まれている場合2点)
- 処方時に後発医薬品の銘柄を記載した上で変更不可とする場合は、処方せんにその理由を記載
平成30(2018)年4月
平成30(2018)年度診療報酬、調剤報酬改定
- 後発医薬品調剤体制加算の段階的評価の見直し(75%以上18点、80%以上22点、85%以上26点)
- 後発医薬品使用体制加算の段階的評価の見直し(85%以上45点、80%以上40点、70%以上35点、60%以上22点)
DPC対象病棟患者の除外規定を廃止、機能評価係数1で評価(下記4) - 外来後発医薬品使用体制加算の段階的評価の見直し(85%以上5点、75%以上4点、70%以上2点)
- DPC病院の機能評価係数2の後発医薬品指数を廃止し、機能評価係数1の評価に組み込む。
- 一般名処方の更なる推進のため、評価点数の引き上げ(後発医薬品が存在する全ての医薬品が一般名処方されている場合6点、1品目でも一般名処方された医薬品が含まれている場合4点)
平成30(2018)年6月
平成30年6月15日、「経済財政運営と改革の基本方針2018~少子高齢化の克服による持続的な成長経路の実現~」(骨太方針)が経済財政諮問会議での答申を経て、閣議決定されました。この中で、「後発医薬品の使用促進についても引き続き取り組む。」と明記されました。
令和2(2020)年4月
令和2(2020)年診療報酬、調剤報酬改定
- 後発医薬品調剤体制加算の段階的評価の見直し(75%以上15点、80%以上22点、85%以上28点)
- 後発医薬品使用体制加算の段階的評価の見直し(85%以上47点、80%以上42点、70%以上37点)
- 一般名処方の更なる推進のため、評価点数の引き上げ(後発医薬品が存在する全ての医薬品が一般名処方されている場合7点、1品目でも一般名処方された医薬品が含まれている場合5点)
拡大するジェネリック医薬品市場
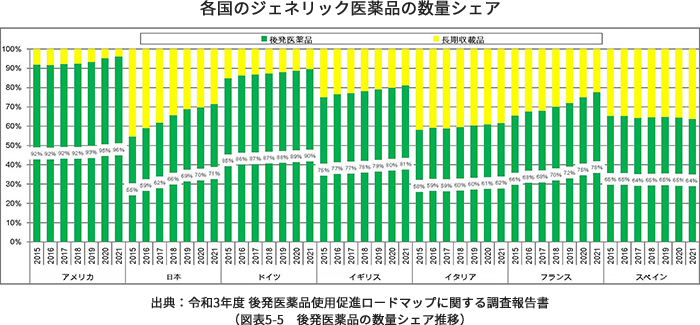
日本でのジェネリック医薬品の使用率は年々伸びておりますが、2019年度(令和元年度)のジェネリック医薬品の数量シェア分析結果では69.0%と世界の使用率に比べると、まだ低い状況にあります。国はこれまで、『2020年度(平成32年度)9月末までに、後発医薬品の使用割合を80%とし、できる限り早期に達成できるよう、更なる使用促進策を検討する』(経済財政運営と改革の基本方針2017)と掲げ、ジェネリック医薬品の使用を進めてきました。現在、国全体では数量シェアはほぼ80%に達しています。一方で、都道府県別に見ると、特に東京、神奈川、大阪などの大都市圏においてまだ未達となっています。日本の国民皆保険制度維持のためには、こうした地域においてもジェネリック医薬品の積極的な使用が期待されることから、引き続き、国はジェネリック医薬品の使用促進を進めています。
Copyright ©2022 IQVIA. IQVIA MIDAS, Market Segmentation, MAT Sep 2015-2021, RX only(PRESCRIPTION BOUND)他をもとに三菱UFJリサーチ&コンサルティング推計無断転載禁止

